
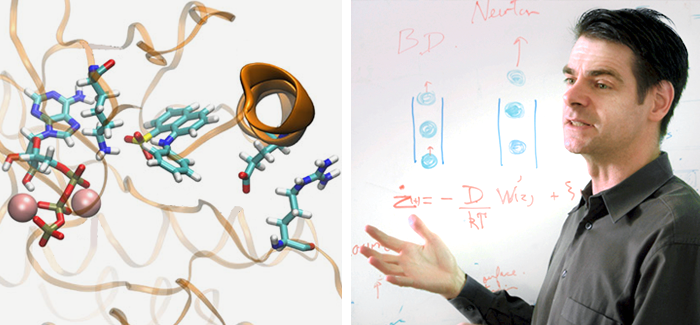
Left: A simulation of Src kinase in midfold, showing one type of chemical bond that can keep the protein inactive. (Illustration courtesy Diwakar Shukla, Yilin Meng, Benoît Roux, and Vijay S. Panda); Right: Roux teaching. (Photography courtesy Benoît Roux)
Simulating a protein that allows cells to run amok, researchers hunt for better cancer drugs.
Researchers at the University of Chicago and Stanford are using the processing power of thousands of idle personal computers—in the homes and offices of ordinary volunteers around the world—to help look for ways to design better, more targeted drugs for cancer treatment.
Cancer is often linked to mutations on enzymes called kinases, which regulate processes such as cell growth. Kinases fold into particular configurations depending on whether they’re active or inactive. Usually cells regulate the kinases and keep them inactive as much as possible. But if the kinases develop mutations that increase their activity, they may induce cells to divide uncontrollably, resulting in cancer. “It’s a bit like the brake of a car,” says Benoît Roux, UChicago professor in biochemistry and molecular biophysics. “If you start to have no brake, then you accelerate more and more and more—it’s going to be bad. So the kinase needs to be able to turn off; it needs to be kept off most of the time. Otherwise you’re really going to be in trouble.”
It’s difficult to develop drugs that inhibit overactive kinases, because scientists don’t know how the molecules transition between inactive and active. “There are lots of crystal structures of kinases in different configurations, druggable or not druggable,” Roux says. “But no one has been able to see how the kinase goes from the off to the on state.”
Roux and his colleagues may have found a way to see these transitions. Using a technique called the string method, the team determined the spatial arrangement of a protein kinase at certain stages as it folds from the inactive to the active state. They passed these snapshots on to Stanford biophysicist Vijay Pande, who teaches structural biology and computer science. Pande used the data to launch computer simulations that allowed the researchers to model how the kinase folds during each small step between active and inactive. “Now we can easily simulate the full transition from the on to the off state and back and forth,” Roux says.
So far the team has performed simulations for a handful of kinases; in a paper this past spring in the journal Nature Communications, Roux and his coauthors reported on Src, a kinase whose activation is associated with many tumors. But there are hundreds more kinase types, Roux says. “And there’s lots of mutations that give resistive effects. For example, a patient could have cancer; you know a given kinase is malfunctioning; you give them a drug; they feel better; and after a while there’s resistance to that drug.” Why does the resistance happen? A new mutation, Roux says. “All the kinases mutated into something else, and now the drug doesn’t bind anymore.” Computer models, he adds, help researchers anticipate the chemical changes that might prevent a drug from binding properly, so that researchers can modify the drug.
Computer simulations are particularly useful for problems such as figuring out the pathways of kinase transitions. Kinases all serve the same function—they remove phosphates from molecules like ATP, which store the energy needed for almost all physiological mechanisms, and attach those phosphates to other proteins. The structural similarities of kinase types make it difficult to develop drugs that target specific kinases and keep them inactive without affecting others. Roux offers an analogy: many doors with the same lock, but you only want to keep one of them closed. “Since they all have the same key, you can’t just focus on the lock,” Roux says. “You have to find something else on the door”—a “novel site”—“to actually jam it.” Despite their common purposes, different kinases fold in unique ways, so once these pathways are established, researchers can pinpoint where drugs bind more easily and “jam” them individually. Then drugs can be tailored accordingly.
Simulations on the Src kinase showed an intermediate state that is “putatively a good place to start to inhibit the protein,” Roux says. “And this intermediate state is not something you normally see.” Now the researchers can simulate the Src kinase’s full transition from the off to the on state.
Running through all the possible ways a kinase can fold takes a large amount of computing power. And the next stage of Roux’s research will involve simulating many more kinases and their interactions with drugs. “You’d like to enter the world of large numbers and maybe simulate a hundred kinases with 15 drugs,” he says. To perform such simulations, the researchers harnessed more than 200,000 idle computers through Folding@home, a Stanford project run by Pande. Ordinary computer users can volunteer their computers by downloading a program from the project’s website that uses the computers’ processing power to run protein-folding simulations while the users are away. “That means that overnight when you’re sleeping,” Roux says, “a drug can run on your computer.”
Computer modeling is an approximation of reality, but one that allows researchers to work with much finer details of a molecule than they otherwise could. Their models, Roux says, serve as imperfect but useful representations. “Ultimately, a drug is a molecule, which binds to a protein, which itself is a macromodel,” Roux says. “If you don’t go to the level of atoms, you’re never going to know what’s going on.”
Video
Computer simulation from the lab of Benoît Roux shows how water molecules physically block the flow of potassium ions through a potassium channel.